.png)
Контроль качества лекарственных средств. Что могут страны?

- Новости /
-
4530
 ЧАСТЬ I
ЧАСТЬ I
Принято говорить, что у каждого врача есть свое кладбище, однако и на работниках фармотрасли лежит ответственность за жизнь и смерть пациентов. Правда, они почти никогда не смотрят в глаза умирающих по их вине больных и их родственников. Хотя… Вот выпуск «New York Times» от 30 апреля 2008 г. [1]. На фотографии — вытирающий слезы немолодой мужчина. Его жена и сын умерли после применения контаминированного препарата гепарина…
Расследование показало, что в Китае на ранних стадиях производства в лекарственное средство (ЛС) был добавлен сульфатированный хондроитин сульфат, который, как и гепарин, обладает антикоагулянтными свойствами, но в 100 раз дешевле в производстве (не 2000, а 20 дол. США за 1 кг). Контроль ЛС на наличие этого вещества не был предусмотрен спецификацией качества на гепарин, и предполагают, что постороннее вещество могло быть добавлено в ЛС намеренно [2]. Итог трагичен — по меньшей мере 81 пациент в США умер по причине побочных реакций на препарат. Другие источники сообщают о более чем 100 смертельных случаях в США и сотнях серьезных побочных реакций в Европе [3] или о более 200 случаев смерти в разных странах [4]. Мощнейшее регуляторное агентство, Управление по контролю за пищевыми продуктами и лекарственными средствами США (Food and Drug Administration — FDA), точнее его Центр по оценке и исследованию лекарственных средств (Center for Drug Evaluation and Research — CDER), который на своей веб-страничке назван сторожевым псом американских потребителей, не защитил этих людей от смертельной опасности. Д-р Джанет Вудкок (Janet Woodcock), директор CDER, отметила по этому поводу на слушаниях в Конгрессе, что было бы лукавством со стороны FDA утверждать, будто оно может инспектировать каждое производство, каждый продукт и что такая инспекция может выявить любую проблему.
FDA не может следить за качеством продуктов на всех этапах их обращения, — это неоднократно подчеркивала Дж. Вудкок [5]. Кроме того, по ее словам, основную ответственность за качество ЛС на разных этапах его обращения несут производители, импортеры, посредники и дистрибьюторы. Уровень требований к ним как со стороны властей, так и широкой общественности будет постоянно расти. Гепариновый кризис, когда к конечному потребителю, пройдя проверку на этапах лекарственной субстанции и готового ЛС, попал препарат, содержащий большое (по массе) количество контаминанта, должен рассматриваться всеми как грозное предупреждение, — отметила директор CDER.
Контроль именно зарубежных производств и экспортируемых препаратов — больной вопрос для управления. После гепаринового скандала его обсуждают особенно активно. Ведь согласно данным Правительственного управления по отчетности (Government Accountability Office) 80% лекарственных субстанций, используемых производителями в США, импортируются, причем преимущественно из Китая [6]. Кроме того, 40% готовых ЛС в США завозят из-за рубежа. Подсчитано, что при существующем положении вещей для проведения разовых инспекционных проверок каждого зарубежного производителя потребуется не меньше 13 лет [7]. Но такое возможно, если бы все оставалось на своих местах. На деле же количество лекарственных продуктов (готовых ЛС, субстанций и вспомогательных веществ), импортируемых в США, как отметила Дж. Вудкок, с 2001 г. более чем удвоилось. То же касается и зарубежных производителей, выпускающих одобренные FDA продукты. Количество инспекций, конечно, увеличивается, но далеко не пропорционально росту числа производителей. Поэтому частота инспекций, как сообщила директор CDER, с 2001 по 2007 г. снизилась на 41% (рис. 1). Система обеспечения качества ЛС, по словам Дж. Вудкок, сегодня стоит перед лицом ряда нерешенных проблем:
|
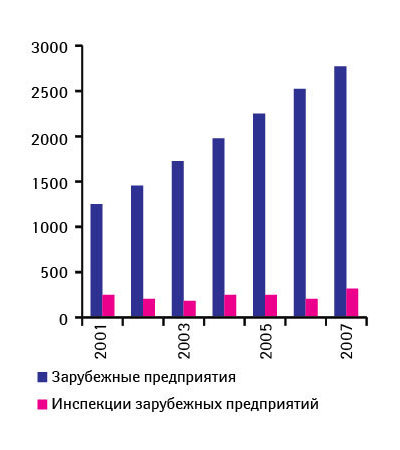
- глобализация производства;
- усиление угроз злоупотреблений и преступлений (фальсификаций, биотерроризма и т.д.);
- продолжающееся разобщение и фрагментированность регуляторной сферы в глобальном масштабе;
- резкое сокращение охвата предприятий инспекциями в последнее время;
- недостаточное внедрение современных информационных технологий (например автоматизированный учет разрешений на маркетинг, лицензий, хода инспекций и т.д.).
Стратегические планы FDA по их решению представлены в ряде программ, в том числе «Фармацевтическое качество ХХI столетию» (Pharmaceutical Quality for the 21st Century). В 2002 г. FDA выступило с инициативой, которая нашла отражение в поистине революционном документе, получившем название «Технологии анализа процессов» (Process Analytical Technology — PAT) [8]. Инициативы РАТ в первую очередь позиционируются как интегрированная система для планирования, анализа и контроля наиболее критических характеристик исходного сырья, компонентов процесса, а также параметров самого процесса с целью обеспечения необходимого качества готового ЛС РАТ предполагает создание системы контроля качества ЛС в реальном времени (то есть в момент выпуска). Один из ключевых элементов РАТ — сделать качество неотъемлемым свойством как самого препарата, так и процесса его производства. Для поддержки инициативы РАТ в CDER создан специальный комитет, разработано руководство. Особое внимание управление уделяет не только качеству, но и безопасности препаратов. «Safety first» (приоритет безопасности) — такую задачу ставит FDA перед отраслью. Управление привлекает новые ресурсы: 600 специалистов были приняты на работу в CDER в 2008 г. В ближайшее время управление рассчитывает нанять еще примерно столько же, так что количество его штатных сотрудников превысит 11 тыс. [9].
Но все же: «Мы не можем быть службой, контролирующей качество, для всего мира», — заявила Дж. Вудкок на вышеупомянутых слушаниях в Конгрессе по поводу инспекций производств, выпускающих за рубежом одобренные FDA продукты. То есть о качестве должен печься прежде всего производитель. Если он не сможет убедить FDA на этапе получения разрешения на маркетинг, ну что же — в США ему путь заказан. Большинства других государств это, к сожалению, не касается. Некоторое представление о проблемах развивающихся стран можно получить, ознакомившись с бюллетенем «Matrix of Drug Quality Reports Affecting USAID-assisted Countries», который готовят в рамках Программы качества ЛС и информации Фармакопеи США (USP Drug Quality and Information Program) [10]. «Согласно источникам из Великобритании в 2003 г. в Китае около 100 000 человек умерли от незаконных ЛС», — сообщается в этом источнике со ссылкой, почему-то, на сайт www.fdanews.com, где к тому же (возможно по причине реконструкции сайта) соответствующую статью обнаружить не удалось. Откуда такая цифра, непонятно, но в том, что субстандартных, фальсифицированных, в том числе смертельно опасных, ЛС в странах третьего мира очень и очень много, сомневаться не приходится. Достаточно хотя бы полистать вышеупомянутый бюллетень.
Многоуровневая система контроля качества ЛС — как компьютерное антивирусное программное обеспечение — нуждается в постоянном совершенствовании. Размещая здесь кое-какую информацию, почерпнутую из общедоступных источников, мы не предполагаем, что специалисты именно в этой сфере найдут здесь для себя что-то новое. Просто маховик государственной системы крутится тогда, когда общество зорко следит за тем, чтобы не было остановок. Поэтому все мы должны знать, и требовать, и участвовать…
ГОСУДАРСТВЕННАЯ СИСТЕМА КОНТРОЛЯ КАЧЕСТВА ЛС США
Система сообщений о качестве ЛС (Drug Quality Reporting System — DQRS) функционирует в США с начала 1970-х годов. В настоящее время сообщения о подозреваемых проблемах качества ЛС посредством телефонной связи или других быстрых коммуникаций (к примеру системы репортирования о побочных реакциях — MedWatch) поступают в Отдел соответствия, управления рисками и надзора CDER (The Division of Compliance Risk Management and Surveillance). Подходы к расследованию случаев детально описаны в презентации «Drug Quality Reporting System» (датирована 18.01.2007 г.) [11]. Соответствующие обязанности разделяют владелец разрешения на маркетинг, региональное подразделение FDA и CDER.
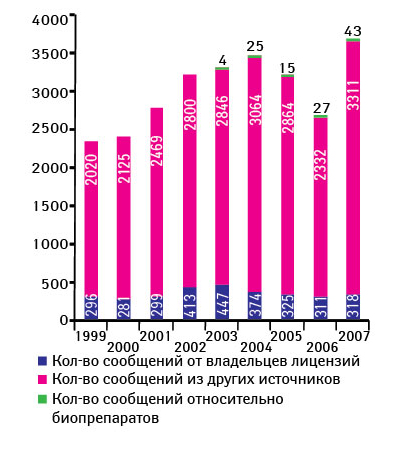
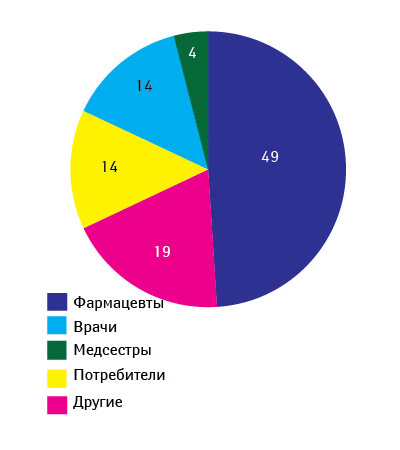
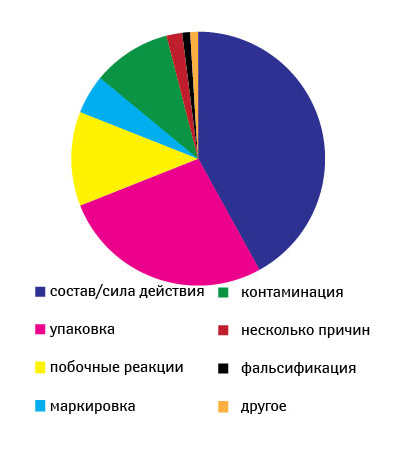
Ежегодно CDER получает около 2,5–3,5 тыс. сообщений о проблемах качества ЛС (период 1999–2007 гг.) (рис. 2) [12]. О возможных проблемах с качеством препаратов от специалистов здравоохранения и потребителей FDA в 2007 г. получило 3311 сообщений, от производителей — 318, а также 43 сообщения, касающихся биопрепаратов. Стоит отметить, что фармацевты отправляют примерно половину сообщений от специалистов здравоохранения и потребителей (рис. 3). В 2007 г. наибольшее количество сообщений было вызвано отклонениями в составе препаратов (42%), состоянии упаковки (27%), побочными реакциями (12%) (рис. 4) [12].
|
|
|
Вообще анализ результатов инспекций производств и проверка качества препаратов, находящихся в обращении, путем отбора и анализа проб — основные стратегические направления надзора FDA за соответствием препаратов установленным стандартам [12]. Выбор препаратов и производств для проверки (как в стране, так и за рубежом) осуществляется на основании оценки рисков. В случае обнаружения отклонений региональные подразделения FDA проводят инспекции для выяснения их причин и разработки корректирующих действий совместно с компанией. Выделяют следующие критерии для отбора проб на основании оценки рисков:
- опасность микробного/эндотоксинового заражения;
- проблемы стабильности;
- проблемы стерильности;
- отклонения по результатам теста растворения;
- превышение допустимого уровня примесей/продуктов распада;
- «скомпрометированная» история препарата;
- подозрение на фальсификацию;
- прежние случаи нарушений.
Рецептурные и безрецептурные препараты в США согласно федеральному законодательству должны соответствовать стандартам Фармакопеи США (United States Pharmacopeia — USP), если таковые существуют. Кстати, USP — организация неправительственная, неприбыльная, в которой работают эксперты-волонтеры в соответствии со строгими правилами, предупреждающими конфликт интересов. Несоответствие компендиальным стандартам качества, силы действия или чистоты, выявленное при тестировании предписанным USP методом, считается фальсифицированным, если в инструкции по медицинскому применению эти отличия четко не обозначены (Федеральный закон о продуктах питания, лекарственных и косметических средствах (Federal Food, Drug, and Cosmetic Act). Если же ЛС не описано в официальном компендиуме, фальсифицированным оно считается в случае несоответствия стандартам качества, указанным в инструкции, спецификациях производителя или заявке на получение разрешения на маркетинг. Согласно закону таковым оно будет считаться, если отклонения выявлены любым научно обоснованным методом [13].
В 2007 фискальном году FDA провело 1119 пострегистрационных внутренних (в результате была приостановлена работа 1 предприятия и выпущено 14 предупредительных писем) и 289 предрегистрационных инспекций (проводятся в обязательном порядке при подаче заявок на получение разрешения на маркетинг оригинальных или генерических препаратов), а также 333 зарубежные инспекции [12].
Интересно, что по закону FDA может своим решением отзывать биопрепараты и медицинское оборудование, но не ЛС, и большинство отзывов проводят по инициативе производителей. Это случается, когда либо само предприятие обнаружило дефекты (нарушения, отклонения), либо их выявило FDA и рекомендовало отзыв. При этом обычно компания следует рекомендации. В противном случае FDA может требовать наложения санкций в предусмотренном Федеральным законом о продуктах питания, лекарственных и косметических средствах порядке. На ЛС может быть наложен арест, против компании выдвинуто обвинение, а препарат может быть отозван по решению суда [14; 15].
Динамика количества отзывов (recalls) (применяют в отношении нескольких серий) за год, отмечает CDER, непоказательна, поскольку рекорд 2007 г. вызван 670 отзывами со стороны одного упаковщика, так что общее количество отозванных рецептурных препаратов составило 851, безрецептурных — 136 [12].
Основными причинами отзыва ЛС с рынка были следующие:
- несоответствие требованиям к упаковке;
- нарушение температурных условий хранения;
- уменьшенное количество действующего вещества;
- химическая контаминация;
- превышение допустимого уровня примесей/продуктов распада;
- несоответствие требованиям теста растворения;
- повреждение маркировки;
- маркетинг в отсутствие одобренной заявки на получение разрешения на маркетинг;
- недостатки в обеспечении стерильности;
- приводящее к ошибкам сходство маркировок.
Последний пункт особенно обращает на себя внимание, так как у нас подобное практически никогда не становилось причиной запрещения обращения ЛС. В США же сходство маркировки с таковой у жизненно важного препарата становится причиной отзыва I класса [16]. Стоит напомнить, что отзыв I класса, как и II, осуществляется до уровня аптек включительно, кроме того, предусматривает обязательное извещение всех пациентов, получающих ЛС.
Удалено с рынка (withdraw) в связи с проблемами безопасности в 2007 г. всего 2 препарата — перголид и тегасерода малеат [12].
По инициативе FDA USP приняло ряд изменений к монографии на гепарин. Первый пересмотр был произведен в феврале 2008 г. с введением новых тестов (при помощи электрофореза и ядерно-магнитного резонанса) для идентификации сульфатированного хондроитин сульфата. Исчерпывающая модификация этой статьи с включением методов и технологий, уже широко используемых производителями, должна вступить в силу в августе 2009 г. [4].
Информации непосредственно о лабораторных проверках маловато, но, в частности, удалось выяснить, что частота отклонений нормативных показателей при этом составляет около 2%. Интересно, что аналогичный показатель у изготовляемых по индивидуальному требованию ЛС составил 34% (по результатам ограниченной проверки, проведенной FDA в 2001 г. [17]).
О количестве ЛС, тестируемых FDA за год, не удалось найти информации. Вот только сообщают, что с 1996 по 2001 г. проанализировано свыше 3000 препаратов разных производителей [17].
О ЧЕМ СООБЩАЕТ БРИТАНСКИЙ РЕГУЛЯТОР?
Зато Агентство по регулированию лекарственных средств и продуктов для здравоохранения Великобритании (Medicines and Healthcare Products Regulatory Agency — MHRA) сообщает на своем сайте, что в рамках программы проверки ЛС (Medicines testing scheme — MTS) ежегодно отбирают и анализируют 2000–2500 образцов препаратов [18]. Целью программы является подтверждение качества ЛС, представленных на рынке Великобритании. Препараты для анализа могут быть взяты либо по требованию других подразделений MHRA, либо в рамках национальной программы надзора за ЛС. По требованию могут отбирать пробы в рамках расследований, инспекций, плановых мероприятий постмаркетингового надзора, перед выдачей разрешений на маркетинг (таблица) [19]. По инициативе сотрудников MTS пробы на этапах производства, хранения, дистрибьюции или розничной продажи отбирают инспекторы Королевского фармацевтического общества (Royal Pharmaceutical Society). Это могут быть новые химические вещества (New chemical entity — NCE), генерики, не получающие разрешение на маркетинг специальные ЛС (см. ниже) и препараты, получившие разрешение на маркетинг по централизованной процедуре.
Таблица 1 | Источники образцов препаратов, проверенных в рамках системы MTS |
Источники | 2004/05 | 2005/06 | 2006/07 | 2007/08 |
На основании DMRC | 44 | 44 | 70 | 83 |
Инспекторат по ЛС | 26 | 16 | 27 | 25 |
Плановые проверки | 54 | 552 | 619 | 406 |
Предрегистрационные | 75 | 194 | 225 | 163 |
Надзор за продуктами на рынке | 2273 | 2055 | 1363 | 1347 |
ЕМЕА*-централизованная процедура | 17 | 16 | 15 | 9 |
Другие | 64 | 31 | 35 | 176 |
Для подтверждения соответствия спецификациям положено в течение первых двух лет маркетинга проанализировать 3 серии новых препаратов, относящихся к NCE. Принято также ежегодно на основании анализа рисков производить отбор и исследование ряда генерических препаратов [20]. С 2002 г. MHRA располагает собственной лабораторией.
Для подтверждения минимальных требований в отношении спецификаций согласно Британской Фармакопее** анализируют не получающие разрешение на маркетинг (специальные) ЛС, изготовленные по специальной лицензии для удовлетворения специфических нужд конкретных пациентов. В период 2000–2005 гг. 56% из 79 таких препаратов оказались несоответствующими регуляторным требованиям. У получивших разрешение на маркетинг препаратов частота отклонений ниже. Правда, большинство выявленных недостатков у не получающих разрешение на маркетинг ЛС не влияли на их качество и безопасность и заключались в нарушениях маркировки, спецификаций и контроля качества производителем. На основании результатов MTS предпринимают более обширные расследования. Их приоритетными направлениями называют:
- выявление источников фальсифицированных ЛС;
- выявление в составе растительных препаратов рецептурных ЛС (глибенкламид, фенфлурамин, сибутрамин, сильденафил, кортикостероиды, дисульфирам, тадалафил);
- выявление в составе растительных препаратов запрещенных или неразрешенных компонентов (эфедра, аристолохия, гидрохинон);
- экспертиза диетических добавок (для уточнения принадлежности продуктов к этой группе);
- токсические примеси в растительных продуктах (тяжелые металлы, нитрозофенфлурамин).
Производители, импортеры и дистрибьюторы обязаны информировать MHRA о любых заподозренных дефектах качества ЛС, которые потенциально требуют отзыва или приостановления поставок. Информирование о возможных дефектах качества ЛС входит также в обязанности специалистов здравоохранения. Потребителям в аналогичных ситуациях MHRA советует по возможности для начала проконсультироваться с врачом или фармацевтом; но если такая возможность отсутствует — сообщать непосредственно в агентство.
В 2007/2008 финансовом году MHRA получило 465 сообщений о проблемах качества ЛС и в поддержку действий производителей, отзывающих препараты или их отдельные серии, издало 34 срочных предупреждения, из которых 8 — класса I (требующие немедленных действий со стороны участников обращения ЛС), 14 класса II (действия в течение 48 ч), 2 класса III (действия в течение 5 дней) и 1 — класса IV (о необходимости осторожного применения). Одно из предупреждений извещало об отзыве препарата, еще пять — о фальсифицированных препаратах [19].
ПОВТОРЕНИЕ ГЕПАРИНОВОЙ ТРАГЕДИИ ВОЗМОЖНО?
Усилия, прикладываемые регуляторными органами для выявления субстандартных и фальсифицированных ЛС, главный специалист по науке USP Даррелл Абернети (Darrell Abernethy) сравнивает с работой Олимпийского комитета по выявлению случаев употребления допинга спортсменами. Ясно, что пока комитет ищет подход к распознаванию одного запрещенного препарата, на свет появляется следующий, выявлять который еще предстоит научиться, — отмечает Д. Абернети. По мнению некоторых экспертов, мы стоим на пороге других страшных последствий применения субстандартных ЛС. Чтобы этого не случилось, предприятия отрасли должны взять на себя всю полноту ответственности за производственный цикл, начиная с сырья для выпуска ЛС, ибо, как с грустной иронией говорит Уоррен Перри (Warren Perry), эксперт из «Qumas Consulting» (Сан-Франциско): «Пока производители греются у очага регуляторов, ничего не изменится» [21]. Когда же отрасль предпримет решительные меры, чтобы минимизировать опасность наступления очередного кризиса? К сожалению, прежде может понадобиться еще несколько тяжелых уроков, цена которых — жизни людей.
Дарья Полякова
Источник: apteka.ua
*Европейское агентство по лекарственным средствам (European Medicines Agency — EMEA);
**всемирно известная Британская Фармакопея является единственным в стране полным собранием стандартов на ЛС в любую фазу их жизненного цикла.
ЛИТЕРАТУРА
- Harris G. Heparin Contamination May Have Been Deliberate, F.D.A. Says. // New York Times. April 30, 2008. Available at: http://www.nytimes.com/2008/04/30/health/policy/30heparin.html?_r=2.
- Committee on Energy and Commerce Subcommittee on Oversight and Investigations. The heparin disaster: Chinese counterfeits and American failures. April 29, 2008. Available at: http://energycommerce.house.gov/cmte_mtgs/110-oi-hrg.042908.Heparin.shtml.
- Annual report of activities of the EDQM — 2008. Available at: http://www.edqm.eu/medias/fichiers/Annual_Report_Activitie.pdf.
- The Standard. 2009. Vol. 6, Issue 3. Available at: http://www.usp.org/pdf/EN/aboutUSP/theStandard2009Winter.pdf.
- Janet Woodcock. Director, Center for Drug Evaluation and Research, FDA. CDER Priorities for 2009. Available at: http://www.fda.gov/downloads/AboutFDA/CentersOffices/CDER/ucm117684.pdf.
- Drug Safety: Preliminary Findings Suggest Weaknesses in FDA’s Program for Inspecting Foreign Drug Manufacturers, GAO-08-224T, November 1, 2007. Available at: http://www.gao.gov/new.items/d08224t.pdf.
- It’s Still the Economy, Stupid. Eagle ForumVol. 41, No. 7 February 2008. Available at: http://www.eagleforum.org/psr/2008/feb08/psrfeb08.html.
- OPS Process Analytical Technology —(PAT) Initiative . Available at: http://www.fda.gov/AboutFDA/CentersOffices/CDER/ucm088828.htm.
- Vivian J.C. FDA Inspection of Foreign Drug Companies US Pharmacist. 2008;33(6):53-57 6/19/2008. Available at: www.uspharmacist.com/content/d/pharmacy_law/c/9789.
- Matrix of Drug Quality Reports Affecting USAID-assisted Countries By the U.S. Pharmacopeia Drug Quality and Information Program. Created by: Joyce Primo-Carpenter, M.D., BSc. Pharm. USP DQI Associate Director, 2003–2008. Updated: June 1, 2009.
- Johnson J. Drug Quality Reporting System Presentation (updated 1/18/2007). Available at: http://www.fda.gov/downloads/AboutFDA/CentersOffices/CDER/ucm102838.pdf.
- CDER 2007 Report (update): Improving Public Health Through Human Drugs. Available at: http://www.fda.gov/downloads/AboutFDA/CentersOffices/CDER/WhatWeDo/UCM121704.pdf.
- CPG Sec. 420.100 Adulteration of Drugs Under Section 501(b) and 501(c) of the Act. Direct Reference Seizure Authority for Adulterated Drugs Under Section 501(b) (CPG 7132a.03) Available at: http://www.fda.gov/ICECI/ComplianceManuals/CompliancePolicyGuidanceManual/ucm074367.htm.
- Federal Food, Drug, and Cosmetic Act (FD&C Act). Available at: http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/default.htm.
- Public Health Service Act. Available at: http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/default.htm.
- Debra B.F. Pharmacy law. McGraw-Hill Professional. 2007.
- Report: Limited FDA Survey of Compounded Drug Products. Available at: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/PharmacyCompounding/ucm155725.htm.
- Medicines testing. Available at: http://www.mhra.gov.uk/Howweregulate/Medicines/Inspectionandstandards/Medicinestesting/index.htm.
- MHRA annual statistics 2007/08. Available at:http://www.mhra.gov.uk/Publications/Corporate/AnnualReports/CON020817.
- Lee G, Charvill A, Heddell G. The MHRA medicines testing scheme: working to protect the public. Pharmaceutical Journal. 2005.Vol. 275.
- Shanley A, Thomas P., Vaccarello M., Ciurczak E. Lessons from Heparin Available аt: http://www.pharmamanufacturing.com/articles/2008/123.html?page=print.
ЧАСТЬ II
Почему-то недавние резонансные события (по большей части — трагедии), связанные с негативными последствиями применения лекарственных средств (ЛС), в Старом и Новом Свете происходили по закону парных случаев: в США — сердечно-сосудистые осложнения после Vioxx, в Европе — суициды на фоне приема антидепрессантов; в США — смертельные случаи в связи с контаминацией препарата гепарина, в ЕС — переполох по поводу контаминированного Viracept. В первой части публикации, посвященной контролю качества ЛС в мире, описаны некоторые обстоятельства гепариновой трагедии [1], которую, не вдаваясь в подробности, эмоционально можно интерпретировать так: «Опять в Китае что-то напортачили, а людям — горе». Но «парный» случай, произошедший в Европе, заставляет задуматься: на этот раз контаминация произошла не на малоизвестном производстве в далеком Китае, а в Швейцарии, у весьма именитого производителя. Как же так: «швейцарское» качество, которым обычно так гордятся, было поставлено под сомнение?
После проведенного расследования Комитет по лекарственным средствам для применения у человека (Committee for Medicinal Products for Human Use — СНМР) дал производителю довольно резкую оценку, заявив о недостаточном знании и понимании производственного процесса препарата Viracept [2]. Знакомство с отчетом СНМР вызывает удивление и недоумение: как вообще такое могло произойти у производителя с мировым именем на самом что ни на есть европейском предприятии? Напомним, что препарат к тому же зарегистрирован по централизованной процедуре, поэтому за его качество отвечают совместно швейцарский регулятор Swissmedic и Европейское агентство по лекарственным средствам (European Medicines Agency — EMEA). Но, как говорится, у двух нянек — дитя без глаза.
Коротко — о дефектах, которые привели к контаминации, согласно вышеупомянутому отчету. 5 июня 2007 г. компания «Roche Registration Limited», владелец разрешения на маркетинг препарата Viracept, сообщила ЕМЕА о контаминации препарата веществом с генотоксическими свойствами и отозвала препарат с рынков (в том числе украинского*).
Этилмезилат (этиловый эфир метансульфоновой кислоты) был выявлен компанией «Roche» в нескольких сериях препарата после жалоб пациентов (6 больных из 3 стран) на странный запах и побочные явления — тошноту и рвоту (у 2 пациентов) [3]. На тот момент степень риска для пациентов была неизвестной, тогда как генотоксические, канцерогенные и тератогенные свойства этилмезилата по отношению к животным были известны. Еврокомиссия, по инициативе ЕМЕА, приостановила действие разрешения на маркетинг Viracept.
Через несколько дней после заявления «Roche» Swissmedic совместно с ЕМЕА организовали инспекцию предприятия, а еще меньше чем через неделю ее результаты были представлены СНМР [2]. За этим последовал ряд встреч и заседаний экспертов, а через 3 мес — повторная инспекция. На основании ее результатов, с учетом ответов на вопросы, полученных от «Roche», СНМР принял решение восстановить действие разрешения на маркетинг Viracept.
Как же произошла контаминация? Оказалось, что GMP была нарушена на уровне резервуара, в котором хранили метансульфоновую кислоту, используемую на последнем отрезке фармацевтического синтеза для преобразования нелфинавира основного в нелфинавир мезилат. Этот резервуар не был включен в стандартную операционную процедуру (СОП) очистки оборудования, и его вообще не мыли с 2001 г. до окончания производственной кампании в апреле 2006 г. Затем емкость вымыли согласно СОП (то есть этанолом), но не просушили (!). После этого емкость, в которой были остатки спирта, снова загрузили метансульфоновой кислотой; произошла реакция с образованием этилмезилата в большом количестве. К сожалению, метансульфоновую кислоту рутинно проверяли на наличие этилмезилата только непосредственно по получении от поставщика, а не после загрузки в резервуар на хранение. Контаминированную метансульфоновую кислоту использовали для производства в следующую кампанию — в октябре 2006 г. Препарат трех выпущенных тогда серий, как оказалось впоследствии, содержал повышенное количество этилмезилата (от <1‰ до 8‰). Оставшееся сырье хранилось до следующей кампании, стартовавшей в январе 2007 г., и вследствие длительного хранения в резервуаре образовалось еще больше этилмезилата — до 2300‰.
Этот основной источник контаминации быстро выявила сама компания, однако и в образцах из серий, выпущенных до октября 2006 г., уровень этилмезилата был повышен. По данным компании, в отдельных сериях он достигал 25‰ в период 1999–2003 гг. и 132‰ в период 2003 — март 2007 гг. [4]. Однако при производстве конечного продукта концентрация примеси снижалась примерно на 60%, так что наибольший уровень контаминанта в готовом продукте (в марте 2007 г.) достигал 920‰.
В ходе инспекции оказалось, что, кроме вышеописанной причины, контаминация была вызвана еще двумя: (1) конструкцией трубопровода для подачи этанола и азота, допускавшей вариабельность поступающего в резервуар количества этанола; (2) примесь метил метансульфоната в метансульфоновой кислоте могла медленно трансформироваться в этилмезилат в ходе реакции трансэтерификации [2].
Причиной нарушения качества более общего характера, по заключению СНМР, является то, что компания приобрела права на производство; и поскольку ею не выполнялась фармацевтическая разработка, не был распознан потенциальный риск образования этилмезилата.
Способность метансульфоната в присутствии этанола образовывать этилмезилат описана в компендиальных статьях о действующих веществах в форме мезилата (например статье на нелфинавира мезилат Международной Фармакопеи). Более того, в 2001 г. СНМР напоминал об этой проблеме владельцу разрешения на маркетинг нелфинавира мезилата. СНМР в своем отчете напоминает также, что предельный уровень этилмезилата сейчас установлен при показателе 0,5‰, а также что общим принципом недопущения присутствия примесей в фармацевтической субстанции является проверка реагентов перед их использованием.
«Roche» ответила регулятору, что проблема, вызвавшая контаминацию, ясна, и не составляет большого труда ее разрешить [5]. Почему же производитель взялся за это только сейчас?
Viracept — верхушка айсберга
Вряд ли инспекторы открыли глаза производственникам, выпускающим препарат с 1998 г., на причины образования этилмезилата. Как будет показано ниже, они, вероятно, знали о том, что эта примесь возникает, но в какой-то момент потеряли бдительность и недооценили фактор времени: чем дальше, тем больше образуется контаминанта. Как сообщает in-PharmaTechnologist.com, новостное интернет-издание, от представителя «Roche» стало известно, что в компании хорошо знали о присутствии в Viracept небольшого фонового уровня этилмезилата, образующегося в результате реакции между действующим веществом и алкоголем, остающимся после мойки. На вопрос, почему же компания не изучила, насколько вредно это генотоксическое вещество для пациентов, представитель компании сказал просто, что регуляторный процесс выполнялся адекватно.
Оказывается, ситуация еще более запутанная, и «Roche» действовала просто, «как все», но только ошибка персонала подняла проблему на совершенно другой уровень. В связи с этим свое отношение к примеси этилмезилата в ЛС пришлось публично выразить регуляторным органам по обеим сторонам Атлантики. Только американский регулятор занял гораздо более спокойную позицию и не стал отзывать Viracept, который производит и представляет на рынке США (а также Канады и Пуэрто-Рико) компания «Pfizer». Нужно отметить, что поначалу фармацевтический гигант заявлял о применении им специальных предупредительных мер в отношении контаминации этилмезилатом, а Управление по контролю за пищевыми продуктами и лекарственными средствами США (Food and Drug Administration — FDA) воздерживалось от разъяснений относительно регулирования наличия этой примеси в ЛС [6]. Несколько месяцев спустя управление обратилось к «Pfizer» с просьбой разработать и внедрить новые спецификации относительно содержания этилмезилата в препарате Viracept [7]. Но прежде американский производитель протестировал все активные фармацевтические ингредиенты, и уровень этилмезилата в них оказался гораздо ниже, чем в отозванном продукте «Roche». «Pfizer» и FDA согласовали два вида спецификаций: промежуточная (действовала на протяжении 6 мес) ограничивает повышение риска развития онкозаболеваний у взрослых на протяжении жизни на уровне 17 случаев на 100 000 пациентов; долгосрочная — менее 1 случая (средняя фоновая частота развития онкозаболеваний у пациентов с ВИЧ составляет 20–30 случаев на 1000 пациенто-лет). Больше того, FDA разработало руководство для фармотрасли, посвященное проблеме уменьшения содержания в ЛС подобных примесей [8]. По словам Джеффа Мюррея (Jeff Murray), заместителя директора отдела по противовирусным продуктам Центра по оценке и исследованию лекарственных средств США (Center for Drug Evaluation and Research), этилмезилат и подобные ему генотоксические вещества содержатся во многих ЛС, как уже зарегистрированных, так и находящихся в разработке [9]. Оказывается, и FDA, и ЕМЕА уже несколько лет озабочены этой проблемой и заняты разработкой рекомендаций для отрасли. Самое сложное в этом — определить допустимый уровень содержания в ЛС примесей с генотоксическими свойствами, и случай с Viracept только способствовал активизации обсуждения вопроса [10]. Между тем ЕМЕА, как выяснилось, еще с 2002 г. разрабатывало руководство по генотоксическим примесям [11]. В общем «Roche» просто не повезло первой обнаружить проблему, которую ранее широко не обсуждали.
| ||||||
А насколько, грубо говоря, не повезло пациентам, принимавшим контаминированный препарат? По данным «Roche», около 45 тыс. пациентов применяли Viracept на момент отзыва. По приблизительным оценкам, 20–30 тыс. пациентов были из стран, куда поставляли препарат с высоким содержанием контаминанта (>1000‰ по действующему веществу) [4]. Проведя исследования in vivo, компания показала возможность определить пороговую величину поступления в организм этилмезилата, ниже которой он не вызывает необратимых мутаций ДНК [12]. По заключению СНМР, пациенты или дети, рожденные от них, подвергались воздействию этилмезилата в количестве меньше порогового, поэтому риск развития онкозаболеваний у них не повысился. На этом основании СНМР заключил, что необходимость в имплементации программ мониторинга пациентов, принимавших контаминированный Viracept, отсутствует.
Объединенная Европа и качество ЛС
Выше было отмечено, что за качество зарегистрированного по централизованной процедуре препарата несут совместную ответственность ЕМЕА и национальные регуляторные органы. Что это значит? Дело в том, что с 1995 г. тесное сотрудничество регуляторных органов стран — членов ЕС и Европейского Экономического Сообщества, в том числе взаимное признание результатов экспертизы, стало обычным явлением при регистрации ЛС [13]. Затем этот опыт распространился на другие сферы деятельности. Одна из них — постмаркетинговый надзор препаратов, получивших разрешение на маркетинг по централизованной процедуре. Этим препятствуют дублированию деятельности, размыванию ответственности и неэффективному использованию ресурсов.
В ЕС обеспечение качества ЛС осуществляется под эгидой Европейского директората качества лекарственных средств и медицинской помощи (The European Directorate for the Quality of Medicines&HealthCare — EDQM). К примеру, в 2008 г. EDQM начато несколько исследований рынка (market surveillance studies), целью которых является скрининг качества ЛС, а именно: лизиноприла дигидрата в форме таблеток; суспензии для интрамаммарного введения, содержащей амоксициллин, клавулановую кислоту и преднизолон; омепразола в форме растворимых в желудке таблеток и капсул [14]. В 2008 г. было проведено первый раз еще одно интересное исследование: 19 лабораторий получили задание определить действующее вещество 4 неизвестных препаратов в форме таблеток любым доступным им методом. Все лаборатории успешно справились с заданием.
Надзор качества ЛС, зарегистрированных по централизованной процедуре
Как известно, на рынок стран — членов ЕС ЛС попадают после получения ими разрешения на маркетинг по одной из процедур [15]:
- централизованной;
- децентрализованной;
- взаимного признания;
- национальной.
По централизованной процедуре обязательно регистрировать ЛС, синтезированные одним из биотехнологических методов [16]:
- рекомбинантной ДНК;
- контролируемой экспрессии генов, кодирующих биологически активные протеины;
- гибридом и моноклональных антител.
С ноября 2005 г. такую процедуру в обязательном порядке проходят также препараты, предназначенные для применения при следующих заболеваниях:
- сахарный диабет;
- онкологические заболевания;
- синдром приобретенного иммунодефицита;
- нейродегенеративные расстройства (в том числе болезнь Альцгеймера);
- препараты-сироты.
Такая процедура предусмотрена и для генерических копий зарегистрированных по централизованной процедуре биотехнологических препаратов. Желательно, но не обязательно, регистрировать таким образом ЛС, относящиеся к NME (new molecular entity — действующее вещество, препараты с которым никогда не получали разрешения на маркетинг), и еще те, что представляют собой значительную медицинскую, научную или технологическую инновацию.
Заявку на получение разрешения на маркетинг по централизованной процедуре подают в ЕМЕА [15]. Один из консультативных комитетов назначает докладчика и содокладчика, рассматривающих досье и готовящих проект отчета; другие члены комитета готовят свои комментарии. В результате появляются заключение комитета, финальный оценочный отчет и краткое резюме характеристик продукта.
Ответственность за мониторинг качества ЛС, получивших разрешение на маркетинг по централизованной процедуре, ныне разделяют регуляторные органы стран — членов ЕС и ЕЭС. ЕМЕА согласно постановлению (ЕС) № 726/2004 вменяется в обязанность:
- координировать надзор применения ЛС в рамках одобренных условий;
- координировать надзор качества ЛС на рынке путем лабораторной проверки соответствия спецификациям.
| ||||||
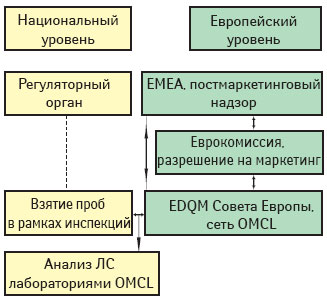
В середине 1990-х годов в Страсбурге (Франция) под эгидой Совета Европы начала функционировать сеть, объединившая Официальные лаборатории контроля качества лекарственных средств (European Official Medicines Control Laboratories — OMCL) Совета Европы. Целью сети является координация административной и технической деятельности OMCL и выработка общих стандартов ЛС. Сотрудничающие с ней регуляторные органы выполняют лабораторный анализ либо посредством своих собственных OMCL, входящих в сеть, либо контрактных лабораторий. В ЕС фармацевтический контроль выполняют государственные лаборатории, финансируемые исключительно за бюджетные средства (включая выплаты за регистрацию и инспекции), и такой их статус гарантирует независимость. Регуляторные органы могут на контрактных условиях использовать услуги частных лабораторий, заключая при этом соглашения о конфиденциальности и отсутствии конфликта интересов [13].
Контрольные лаборатории проверяют образцы ЛС для определения соответствия спецификациям, выработанным во время процедуры выдачи разрешения на маркетинг. Также принимают во внимание методы, описанные в Европейской Фармакопее, других официальных спецификациях и руководствах. С появлением высокотехнологичных инновационных ЛС остро ощущается необходимость разработки и внедрения новых аналитических методик, в чем OMCL также принимают участие.
Общая процедура отбора проб и проверки таких препаратов, основанная на сотрудничестве EMEA, EDQM и национальных регуляторных органов, ЕМЕА внедряет с 1999 г. Согласно этой схеме (схема 1) ЕМЕА несет общую ответственность, а EDQM координирует отбор проб с последующим их анализом.
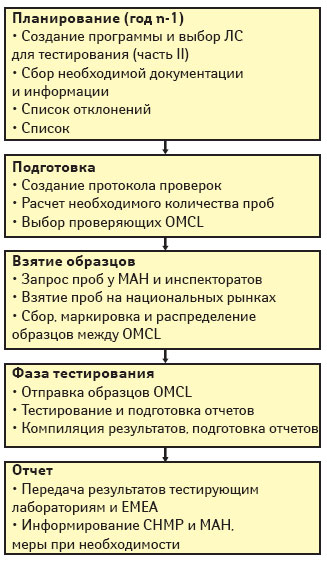
Общая процедура отбора проб и проверки препаратов, получивших разрешение на маркетинг по централизованной процедуре, описана в документе PA/PH/CAP (05) 49 R [17]. Он содержит пошаговое описание работы над программой от планирования (год n-1) до представления годового отчета в ЕМЕА (год n+1).
В январе (года n-1) секретариат ЕМЕА готовит в сотрудничестве с научными комитетами ЕМЕА предложения, базирующиеся на следующих критериях:
1) продукты, получившие разрешение на маркетинг в году n-3 (так называемые автоматические кандидаты, за исключением препаратов, подлежащих посерийному контролю (только биопрепараты));
2) продукты, выбранные на основании анализа рисков и других специфических соображений.
Окончательную версию программы обычно принимают в феврале (года n-1) (схема 2). Вскоре после утверждения окончательного списка проверяемых продуктов ЕМЕА обращается к владельцу разрешения на маркетинг для получения сведений из оригинальной заявки. Компанию просят также проинформировать EDQM о текущей и прогнозируемой рыночной ситуации в отношении продукта и о характере его дистрибьюции в разных странах-членах. Помимо документации из файла заявки, владелец разрешения на маркетинг предоставляет EDQM все релевантные полные действующие СОП лабораторных проверок. Документация хранится в архивной системе с ограниченным доступом. Начиная с весны и заканчивая концом года n-1, выполняют компиляцию всех избранных методов тестирования и готовят его протокол. Каждый протокол содержит информацию о составе препарата, спецификациях и методах проверки плюс методы проверки из досье. К середине октября того же года все протоколы должны быть готовы.
Предварительный план отбора проб составляют на основании сведений о рыночной ситуации. Образцы отбирают на разных этапах сети дистрибьюции уполномоченные национальные службы, обычно трех стран-членов. Выбор стран осуществляется EDQM с тем, чтобы равномерно распределить нагрузку между ними. Размер пробы определяют индивидуально от случая к случаю в зависимости от необходимого для проведения того или иного анализа, силы действия препарата, лекарственной формы, доступности продукта, размера рынка, особенностей клинического использования препарата и т.д. Из каждой страны берут ЛС, принадлежащие только к одной серии для обеспечения сравнимости и адекватности результатов разных тестов. Обычно каждая проба состоит из препаратов 3 серий (по одной из каждой страны).
В августе–сентябре года n-1 EDQM отправляет предварительный план отбора образцов каждому национальному уполномоченному органу, предписывая им в течение месяца подтвердить наличие на их рынках продуктов, о которых запрашивает EDQM. Предварительный план также представляют в сентябре года n-1 в ходе заседания службы GMP-инспекций. В случае если доступность какого-то продукта в одной из стран оказывается под вопросом, обязанность по отбору проб передают другой стране.
После получения ответов самое позднее к ноябрю года n-1 EDQM создает окончательный план отбора проб. Фаза отбора проб должна начинаться к концу года n-1, чтобы начать лабораторные проверки к январю года n.
Запрос относительно аналитических возможностей (testing questionnaire) в виде таблицы содержит параметры тестирования, аналитические методики и специфическое оборудование (в том числе необходимые реагенты), которые требуются для каждого из продуктов. Также в запросе помещают информацию о самом продукте и распространяют в сети OMCL в августе–сентябре года n-1. Лаборатории — потенциальные участницы проверок таким образом получают представление об аналитических методиках и необходимом оборудовании и в течение месяца могут заявить о намерении участвовать в проверке.
Количество лабораторий (OMCL), участвующих в тестировании одного продукта, определяют для каждого случая. Одним из приоритетных критериев для выбора OMCL-кандидатов является доступность технических средств/оборудования (OMCL с системой качества, соответствующей требованиям руководств сети ISO 17025).
Роль и ответственность ЕМЕА
Выбор ЛС для включения в программу ежегодного тестирования выполняется ЕМЕА. Агентство готовит список потенциальных продуктов-кандидатов для тестирования в ближайшие годы и принятие программы научными комитетами. Все сообщения с докладчиками/содокладчиками (ответственными за оценку заявок на получение разрешений на маркетинг и рекомендующими параметры тестирования для программы проверок ЛС) координируются ЕМЕА. ЕМЕА также ответственно за запрос документов и наведение справок у владельцев разрешений на маркетинг о рыночной ситуации с конкретным ЛС. ЕМЕА передает результаты проверок CНMP и связанным с ним рабочим группам, а также владельцу разрешения на маркетинг. Более того, оно регулярно информирует о прогрессе и выполнимости программы. Любые меры на основании результатов анализа ЕМЕА предпринимает в тесном сотрудничестве с EDQM.
Роль и ответственность EDQM
EDQM ответствен за координацию сети OMCL, организацию системы обеспечения качества для OMCL, разработку и поддержание процедур и методов программы отбора образцов и тестирования препаратов. EDQM отвечает за запрос референтных материалов и специфических реагентов у владельца. Он также координирует отбор образцов продуктов с рынков. Выбор OMCL, а также разработка и распространение протоколов проверок выполняется EDQM. После тестирования EDQM собирает все результаты и отчеты и готовит регулярные дополнения о ходе работы программы.
Роль регуляторных органов
На национальные регуляторные органы возложена ответственность взятия образцов продуктов, получивших разрешение на маркетинг по централизованной процедуре, на внутренних рынках по запросу EDQM. Это выполняется их инспекционными службами. Еще одна ответственность — тестирование взятых образцов, выполняемое лабораториями сети.
Ход программы
Взятые образцы рандомизированно направляют в инспекционную службу, обычно каждый препарат трем различным странам — членам ЕС. Количество образцов рассчитывают на основании методов тестирования, рекомендованных докладчиком/содокладчиком и СОП владельца разрешения на маркетинг. Вторым фактором, который принимают во внимание, является необходимость достижения статистической значимости результатов. Образцы берут из нескольких источников в сети дистрибьюции (аптеки, склады, лечебные учреждения).
Наблюдение
препаратов, одобренных странами-членами
Разрешения на маркетинг по процедуре взаимного признания и децентрализованной процедуре выдаются отдельными государствами ЕС. После размещения этих продуктов на национальных рынках ответственность за контроль их качества страны несут сами [18]. Однако заявки на получение разрешения на маркетинг этих продуктов во всех государствах ЕС базируются на идентичном досье, включая идентичные спецификации, что дает возможность создания схемы надзора на уровне ЕС/ЕЭЗ. Для постмаркетингового наблюдения таких препаратов в сети OMCL разработана схема добровольного постмаркетингового надзора.
Надежность результатов тестирования могут лучше обеспечить тестирующие лаборатории с системами качества, которые отвечают требованиям ISO/IEC 17025 и успешно прошли оценку признанного на международном уровне органа или организаций внутри сети OMCL. Внутри сети была разработана основанная на риске модель выбора ЛС для тестирования в рамках постмаркетингового надзора. Эту модель используют все члены сети, не только группа постсертификационного надзора MRP/DCP-продуктов (разрешение на маркетинг которых было выдано по децентрализованной процедуре или процедуре взаимного признания).
Государства ЕС/OMCL, участвующие в схеме надзора MRP/DCP-продуктов, составляют свои планы тестирования, вводя их в качестве предлагаемых проектов в базу данных по тестированию MRP/DCP-продуктов. Планы тестирования, поступающие от всех участников, вместе формируют рабочую программу по регулярному надзору MRP/DCP-продуктов. На основании проектов в этом плане все участники могут планировать отбор образцов продуктов со своего собственного рынка. Результат тестирования сообщается всем участникам посредством базы данных о тестировании MRP/DCP-продуктов, и по запросу полные результаты направляются заинтересованным участникам.
Планы тестирования могут запрашивать в любой момент в режиме онлайн имеющие права доступа пользователи базы данных OMCL. Эти планы обсуждаются участниками во избежание совпадений/повторений и для принятия других организационных мер. Когда продукт находится на рынке реферативного государства ЕС или государства ЕС, в котором осуществляются ключевые этапы производства, тестирование этого продукта в OMCL облегчило бы последующую деятельность, связанную с досье или инспекциями соответственно.
OMCL, идентифицированная как тестирующая лаборатория в рабочей программе по регулярному надзору (или компетентный орган государства ЕС), отбирает образцы продуктов с национального рынка и начинает тестирование.
До окончания срока отбора образцов другие участники (OMCL или их соответствующие компетентные органы) договариваются по поводу размера выборки, параметров тестирования, его графика и т.д., если только это не было определено ранее.
Поощряется отбор образцов в более чем одном государстве ЕС. OMCL тестируют продукты на основании методов, утвержденных владельцем разрешения на маркетинг, или других методов, валидированных согласно принципам обеспечения качества сети OMCL (фармакопейный метод и т.д.).
Как можно скорее после завершения контроля тестирующая OMCL помещает результаты по всем образцам, своим собственным и образцам, направленным другими участниками, в базу данных о тестировании MRP/DCP-продукта в рамках соответствующего проекта. Эти резюмированные отчеты (обобщающие отчеты) являются неотъемлемой частью базы данных, и система генерирует их автоматически.
Вопросы, поставленные во время тестирования, резюмируются тестирующими лабораториями в базе данных тестирования MRP/DCP-продукта под пунктом «Последующие меры». Раздел «Последующие меры» можно добавлять отдельно от введения результатов после завершения контроля и постоянно обновлять.
В основном ответственность за последующие меры лежит на национальных органах. Таким образом, компетентный орган государства ЕС, в котором продукт отбирался в качестве образца, несет ответственность за все последующие меры. Поощряется информирование участниками друг друга в случае необходимости таких мер. В случаях, когда вопрос касается образцов, полученных из нескольких стран, последующие меры следует обсудить между задействованными OMCL-участницами.
Результаты тестирования в соответствии с рабочей программой по регулярному надзору тестирующая лаборатория регулярно вносит в базу данных. Что касается завершенных проектов, то все пользователи базы данных могут автоматически создавать индивидуальные обобщающие отчеты о MRP/DCP-продукте. Участники имеют доступ к данным посредством защищенного портала. Кроме того, доступ (дающий право только на чтение) к базе данных предоставляется по запросу экспертам по качеству, инспекторам и членам групп фармаконадзора национальных компетентных органов государств ЕС/ЕЭЗ.
Дарья Полякова
Литература
1. «Еженедельник АПТЕКА» № 27 (698) от 06.07.2009 г.
2. CHMP assessment report for Viracept. London, 20 September 2007 Doc.Ref.: EMEA/CHMP/492059/2007.
3. Viracept: lessons to be learnt? Anna Lewcock, 14-Jun-2007, Available at http://www.in-pharmatechnologist.com/Processing-QC/Viracept-lessons-to-be-learnt.
4. Miklos Salgo. Clinical Science Leader HIV Roche Viracept Update CARE 5: 5th HIV/AIDS Management Workshop Kampala, Uganda, 2–3 April 2008.
5. Viracept continues to spiral. Anna Lewcock, 08-Aug-2007. Available at http://www.in-pharmatechnologist.com/Processing-QC/Viracept-continues-to-spiral.
6. Viracept saga continues. Anna Lewcock, 25-Jun-2007. Available at http://www.in-pharmatechnologist.com/Processing-QC/Viracept-saga-continues.
7. VIRACEPT® (nelfinavir mesylate) 250 mg, 625 mg tablets, and Powder for Oral Suspension. Important information for prescribers. Pfizer Inc. September 10, 2007. Available at http://www.fda.gov/downloads/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/UCM154880.pdf.
8. Guidance for Industry. Genotoxic and Carcinogenic Impurities in Drug Substances and Products: Recommended Approaches. DRAFT GUIDANCE. FDA. December 2008.
9. Viracept contaminant could be widespread, FDA says. Anna Lewcock, 25-Sep-2007. Available at www.in-pharmatechnologist.com/Processing-QC/Viracept-contaminant-could-be-widespread-FDA-says.
10. Walker Vernon E., Casciano Daniel A., Tweats David J. The Viracept-EMS case: Impact and outlook. Toxicology Letters. Available online 5 April 2009.
11. Guideline on the limits of genotoxic impurities. Committee for Medicinal Products For Human Use (CHMP). London, 28 June 2006. CPMP/SWP/5199/02. EMEA/CHMP/QWP/251344/2006.
12. PRESS RELEASE Studies assessed by the EMEA indicate no increased risk of developing cancer for patients who have taken Viracept contaminated with ethyl mesilate London, 24 July 2008. Doc. Ref. EMEA/CHMP/382256/2008. Available at http://www.emea.europa.eu/humandocs/PDFs/EPAR/Viracept/38225608en.pdf.
13. Paulsen-Sorman U., Wanko R., Spieser J.-M. Market surveillance of centrally-authorised products. Regulatory Affairs Journals Vol 11 No 6 June 2000.
14. Annual report of activities of the EDQM — 2008. Available at: http://www.edqm.eu/medias/fichiers/Annual_Report_Activitie.pdf.
15. Cuddy B. Relationship between receiving authorities and monitoring authorities. EMEA experience. OECD Event, Villa Tuscolana, Frascati (Roma), Italy, 10–11 April 2008.
16. EC/726/2004 Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency (Consolided version: 20/04/2009, Lithuanian Consolidadted version 30/12/2008). http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/vol-1/reg_2004_726_cons/reg_2004_726_cons_en.pdf.
17. CAP Operational Procedure (PA/PH/CAP (05) 49 R). General procedure for sampling and testing of centrally-authorised products (Adopted 09/12/2005).
18. OMCL Network of the Council of Europe. General document. PA/PH/OMCL (06) 116 6R. Co-operation in Post-Marketing Surveillance of MRP/DCP-Products.
*2 июня представительство компании «Ф. Хоффманн-Ля Рош Лтд.» сообщило об этичном отзыве с рынка Украины всех серий препарата Вирасепт (нелфинавир) в форме таблеток и порошка из-за возможного наличия химических примесей в некоторых из них (www.apteka.ua/online/25441/).
ИСТОЧНИК: http://apteka.ua